神経変性疾患

ハンチントン病(HD)の原因は、脳の中にある線条体と呼ばれる大脳基底核のある部位で神経細胞が失われていく進行性(徐々に症状が進んでいく)の神経変性疾患です。大脳基底核の役割としては、運動制御(動きを滑らかにして協調させる機能)、認知機能、感情や動機付けなど様々な機能を司どっています。
中年期(35~50歳)に発症することが多く、症状はゆっくりと進行します。 “ハンチントン病”という名前は、1872年に初めて「遺伝的な舞踏病」としての医学的な報告を行った、アメリカのジョージ・ハンチントン医師の名前にちなんでつけられました。この病気は、かつては「ハンチントン舞踏病」と呼ばれていました。しかし、全身の不随意運動は症状の一つに過ぎず、誤解を招くため、海外では1980年代から、日本国内では、2001年から「ハンチントン病」と呼ばれるようになりました。
日本では2015年に難病法が施行され、ハンチントン病は難病に指定されました(指定難病8)。診断基準を満たし、重症度分類等に照らして病状の程度が一定程度以上の場合に、難病指定医により作成された診断書とともに申請書を都道府県に提出します。そして審査により、特定医療費(指定難病)受給者証が交付されれば、所得に応じて医療費の一部または全部が公費で負担されます。
日本人には100万人に5~6人未満という稀な病気です。国内で医療受給者証を交付されている患者は2017年度末現在900名です 。( 出典:難病情報センター https://www.nanbyou.or.jp/entry/5354)
外国では特に白人(Caucasian) に多く、10万人に4人から10人の割合で存在しているといわれています。
常染色体顕性遺伝病
ハンチントン病を引き起こす遺伝子(IT15またはハンチンチンと呼称されます)は、第4染色体上にあり、CAGの繰り返し配列に異常が認められます。第4染色体の遺伝子の通常(ハンチントン病の症状がない人)の繰り返し配列の長さは26回以下ですが、ハンチントン病の方は36回以上と染色体上の遺伝子配列が伸長しています。この反復配列の数の長さによって発症年齢が決まり、伸長するほど若い時期に発症(表現促進)するといわれています。伸長する原因はまだ解っていません。
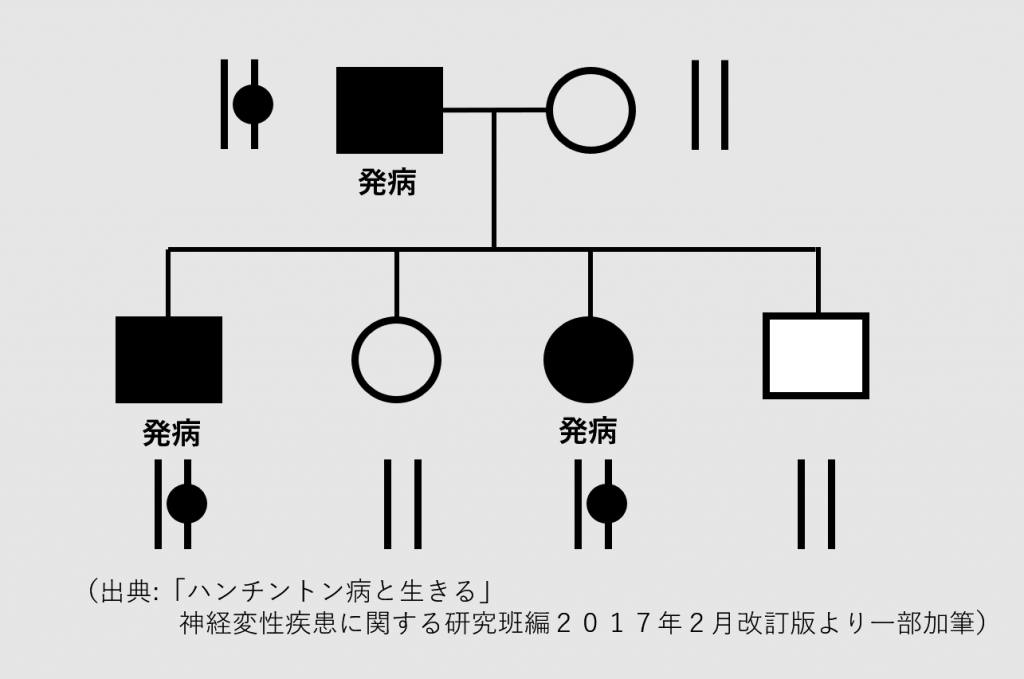
常染色体のため発現と子孫へに伝達については性による影響は受けません。異常遺伝子が子供に遺伝する確率は50%になります。確率が50%ということは2人子供がいれば一人が確実に病気になるということはなく、子供それぞれについて50%の確率で発症するということになります。
現時点では有効な治療薬がありませんので、一つ一つの症状に応じた対症療法しかありません。しかし、科学技術の進歩により、病気の原因となる遺伝子とハンチンチン(huntingtin)というタンパク質の関係を調べる研究や、脳の衰えを和らげるための薬や栄養補助食品の研究、遺伝子治療など、様々な角度から研究が進められています。 また、患者だけではなく、介護者にかかる負担や困難に関する研究のほか、将来、発症する可能性を持つ人たち(at-risk アトリスク)の心理的・社会的問題を調べる研究、どのように家族内でこの病気を分かちあえばよいのかに関する研究も行われています。